The inherited progressive disorder cystic fibrosis (CF) causes severe damage to the lungs, and other tissues in the body by affecting the cells that produce mucus, sweat, and digestive juices. In individuals carrying mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes an ion channel controlling the flow of ions and water in and out of cells, the mucus in airways and other passageways, which normally is thin and slippery, becomes sticky and thick – thus instead of acting as a lubricant, it becomes a congestant.

Advances in patient screenings and breakthrough therapies allow CF patients now to live into their mid- to late 30s or 40s, sometimes even longer. However, even the ~90% of patients (dependent on continent) with the most common CFTR mutation, called ΔF508, who can be treated with available drugs, are still plagued by bacteria settling in their mucus, which causes inflammation in their lungs. The repeated bouts of infection and inflammation, as well as a chronic lower-grade inflammation between infections, gradually weakens and scars patients’ airways, which eventually causes their respiratory systems to fail. For the remaining ~10% of patients with various other CFTR mutations, no targeted treatments even exist yet. A major barrier to developing new and urgently needed treatments is the lack of human in vitro models that recapitulate the CF disease’s pathology.
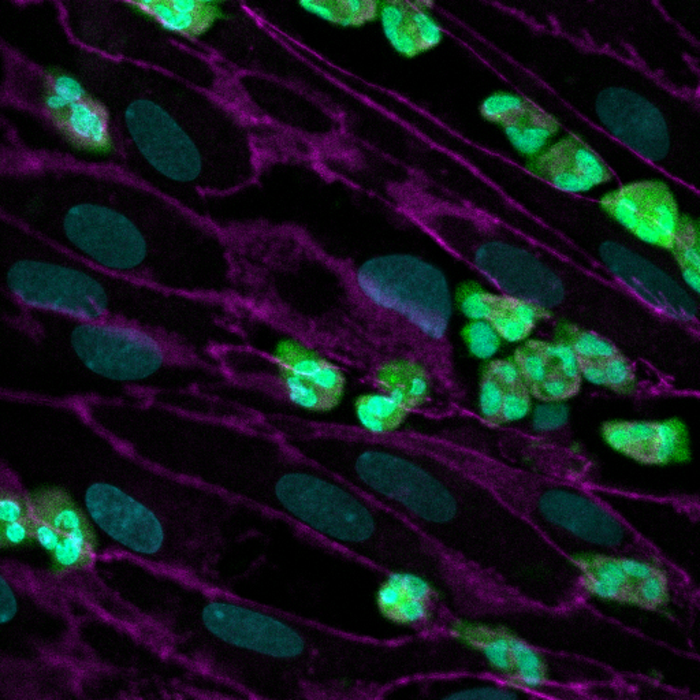
Now, a multidisciplinary research team at Harvard’s Wyss Institute for Biologically Inspired Engineering led by Wyss Founding Director Donald Ingber, M.D., Ph.D. and supported by a grant from the Cystic Fibrosis Foundation, have developed a microfluidic Organ Chip device the size of a USB memory stick that recapitulates key pathological hallmarks from CF patients more accurately than other in vitro systems have so far. The model replicates CF-specific changes in multiple hallmarks of the disease, including in the airway’s mucus layer, beating of mucus-transporting cilia, pathogen growth, inflammatory molecules, and the recruitment of white blood cells, providing a comprehensive preclinical human model in which to investigate new CF therapies. The findings are published in the Journal of Cystic Fibrosis.
“Now that we are able to accurately model CF pathology, including microbiome and inflammatory responses, in human Airway Chips, we have a way to attack challenges that are important to CF patients,” said Ingber. “The bundled capabilities of this advanced in vitro model can help accelerate the search for drugs that may dampen the exaggerated immune response in patients, treat them with more personalized therapies and, help solve problems that CF patients face every day which cannot be addressed by existing treatments.” Ingber also is faculty lead of the Wyss’ Bioinspired Therapeutics & Diagnostics Platform, as well as the Judah Folkman Professor of Vascular Biology at Harvard Medical School and Boston Children’s Hospital, and Professor of Bioengineering at the Harvard John A. Paulson School of Engineering and Applied Sciences
To model and compare the microstructure and function of vascularized CF airways, the authors grew lung airway cells obtained from human CF patients or healthy individuals in one of two parallel running hollow channels of a microfluidic device under air, recapitulating the lung’s air-transporting environment. In the second channel, which is separated from the first one by a porous membrane, they recreated a human blood vessel from human lung microvascular cells that was perfused with a blood substitute medium.
“This first microphysiological model of a CF airway closely mimics what we know from airways in CF patients. Maintaining the typical composition of all relevant cell types, it developed a thicker mucus layer, and its ciliated cells exhibit higher densities of cilia that beat at a higher frequency compared to Airway Chips created with airway cells from healthy individuals,” said co-first author and former Postdoctoral Fellow on Ingber’s team Ratnakar Potla, M.B.B.S., Ph.D. “Importantly, these pathological changes were accompanied by an enhanced inflammatory response in the modeled CF bronchial epithelium that is much like the one observed in CF patients.” Potla is now Senior Scientist of complex in vitrosystems at Genentech-Roche.
After culturing the Airway Chips for two weeks, the researchers measured the levels of pro- and anti-inflammatory factors flowing out of the Airway Chip’s vascular channel that are known to be involved in the inflammatory response in the lungs of CF patients. Among other changes, the level of the pro-inflammatory cytokine IL-8 was increased in CF chips compared to those in control Airway Chips. Interestingly, IL-8 is known for its ability to attract the type of white blood cells know as neutrophils that also drive inflammation in the lungs of CF patients.
When the researchers then actually flowed human neutrophils through the vascular channel of the CF Airway Chips, they observed that more of the immune cells spontaneously adhered to the surface of vascular cells, squeezed themselves through the vascular cell layer and porous membrane, and accumulated in the airway epithelial cell layer of the air channel – recapitulating a process known as “transmigration.”
But the CF-mimicking airway tissue not only stimulated immune cell recruitment, it also supported the growth of the bacterium, Pseudomonas aeruginosa, which is present in the microbiome of normal lung but can grow out of control and cause lung infections in human CF patients. Twenty-four hours after they introduced a fluorescently labeled version of P. aeruginosa into the airway channel of chips created with bronchial epithelial cells from CF patients, the team detected higher numbers of the pathogen in CF-specific mucus than in mucus in healthy Airway Chips. As a consequence, the levels of pro-inflammatory cytokines were further increased, thus replicating the infection-inflammation cycle seen in CF patients.
As next steps, the team will further personalize their CF Lung Airway Chip by generating versions in which bronchial epithelial, vascular endothelial, and immune cells all are obtained from the same patient. “For this proof-of-concept study, we have only used CF-specific bronchial airway cells from patients carrying the frequent ΔF508 CFTR mutation. But different CFTR mutations may also affect the function of endothelial and immune cells and differ in their effects,” said co-first author Roberto Plebani, Ph.D. “By developing a panel of patient-specific Airway Chips using cells from patients with different mutations, and also by directly measuring the activity of differently compromised CFTR ion channels on-chip, drug responses and efficacies could be investigated in a highly personalized manner.” Plebani worked in Ingber’s lab as a visiting professor from the “G. d’Annnunzio” University of Chieti-Pescara in Italy, and spearheaded the project in Ingber’s group with Potla.
IMAGE CREDIT: Wyss Institute at Harvard University



Leave a Reply