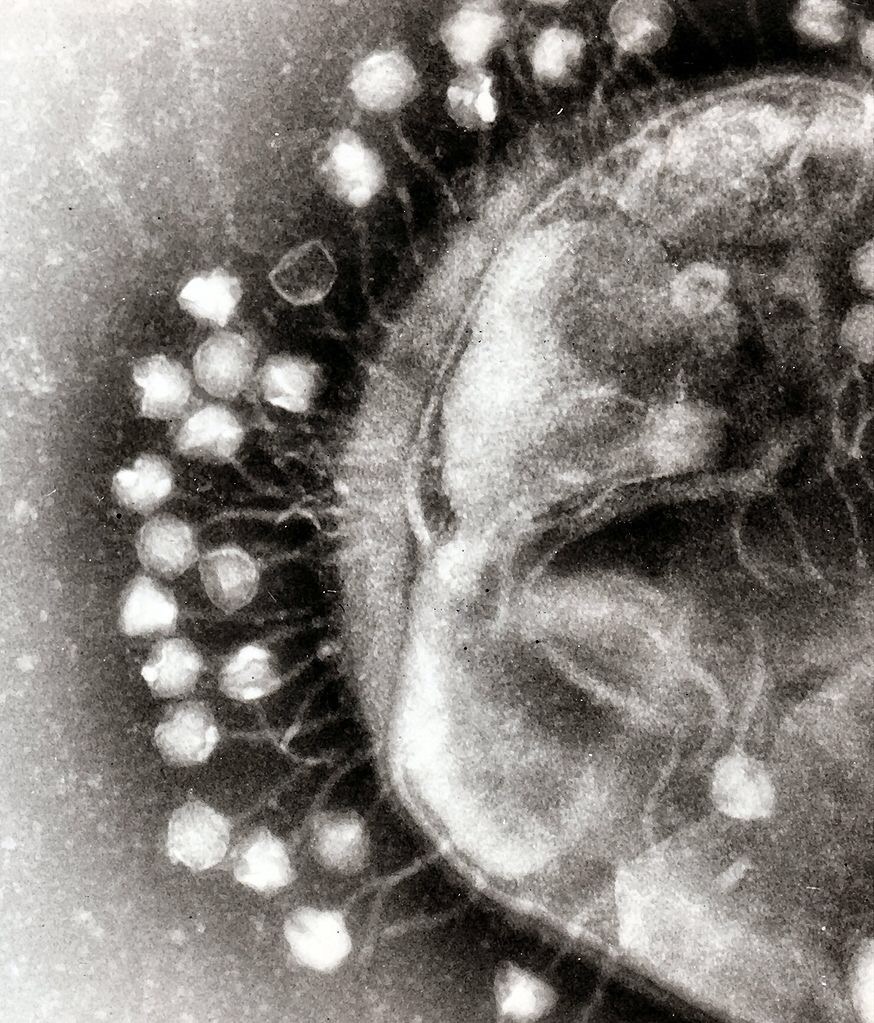
A biological mystery lies at the center of each of our cells, namely: how one meter of DNA can be wadded up into the space of a micron (or one millionth of a meter) within each nucleus of our body.
The nuclei of human cells are not even the most crowded biological place that we know of. Some bacteriophages — viruses that infect and replicate within a bacterium — have even more concentrated DNA.
“How does it get in there?” B. Montgomery (Monte) Pettitt, a biochemist and professor at the University of Texas Medical Branch, asks. “It’s a charged polymer. How does it overcome the repulsion at its liquid crystalline density? How much order and disorder is allowed, and how does this play a role in nucleic acids?”
Using the Stampede and Lonestar5 supercomputers at The University of Texas at Austin’s Texas Advanced Computing Center (TACC), Pettitt investigates how phages’ DNA folds into hyper-confined spaces.
Writing in the June 2017 issue of the Journal of Computational Chemistry, he explained how DNA may overcome both electrostatic repulsion and its natural stiffness.
The key to doing so? Kinks.
The introduction of sharp twists or curves into configurations of DNA packaged within a spherical envelope significantly reduces the overall energies and pressures of the molecule, according to Pettitt.
He and his collaborators used a model that deforms and kinks the DNA every 24 base pairs, which is close to the average length that is predicted from the phage’s DNA sequence. The introduction of such persistent defects not only reduces the total bending energy of confined DNA, but also reduces the electrostatic component of the energy and pressure.
“We show that a broad ensemble of polymer configurations is consistent with the structural data,” he and collaborator Christopher Myers, also of University of Texas Medical Branch, wrote.
Insights like these cannot be gained strictly in the lab. They require supercomputers that serve as molecular microscopes, charting the movement of atoms and atomic bonds at length- and time-scales that are not feasible to study with physical experiments alone.
“In the field of molecular biology, there’s a wonderful interplay between theory, experiment and simulation,” Pettitt said. “We take parameters of experiments and see if they agree with the simulations and theories. This becomes the scientific method for how we now advance our hypotheses.”
Problems like the ones Pettitt is interested in cannot be solved on a desktop computer or a typical campus cluster, but require hundreds of computer processors working in parallel to mimic the minute movements and physical forces of molecules in a cell.
Pettitt is able to access TACC’s supercomputers in part because of a unique program known as the University of Texas Research Cyberinfrastructure (UTRC) initiative, which makes TACC’s computing resources, expertise and training available to researchers within the University of Texas Systems’ 14 institutions.
“Computational research, like that of Dr. Pettitt, which seeks to bridge our understanding of physical, chemical, and ultimately biological phenomena, involves so many calculations that it’s only really approachable on large supercomputers like TACC’s Stampede or Lonestar5 systems,” said Brian Beck, a life sciences researcher at TACC.
“Having TACC supercomputing resources available is critical to this style of research,” Pettitt said.
FINDING THE ORDER IN DISORDERED PROTEINS
Another phenomenon that has long interested Pettitt is the behavior of Intrinsically Disordered Proteins (IDPs) and intrinsically disordered domains, where parts of a protein have a disordered shape.
Unlike crystals or the highly-packed DNA in viruses, which have distinct, rigid shapes, IDPs “fold up into a gooey mess,” according to Pettitt. And yet they’re critical for all forms of life.
It is believed that in eukaryotes (organisms whose cells have complex substructures like nuclei), roughly 30 percent of proteins have an intrinsically disordered domain. More than 60 percent of proteins involved in cell signaling (molecular processes that take signals from outside the cell or across cells that tell the cell what behaviors to turn on and off in response) have disordered domains. Similarly, 80 percent of cancer-related signaling proteins have IDP regions – making them important molecules to understand.
Among the IDPs Pettitt and his group are studying are nuclear transcription factors. These molecules control the expression of genes and have a signaling domain that is rich in the flexible amino acid, glycine.
The folding of the nuclear transcription factor signaling domain is not brought about by hydrogen bonding and hydrophobic effects, like most protein molecules, according to Pettitt. Rather, when the longer molecules find too many glycines in a space, they go beyond their solubility and start associating with each other in unusual ways.
“It’s like adding too much sugar in your tea,” Pettitt explains. “It won’t get any sweeter. The sugar must fall out of solution and find a partner – precipitating into a lump.”
Writing in Protein Science in 2015, he described molecular simulations performed on Stampede that helped to explain how and why IDPs collapse into globule-like structures.
The simulations calculated the forces from carbonyl (CO) dipole-dipole interactions — attractions between the positive end of one polar molecule and the negative end of another polar molecule. He determined that these interactions are more important in the collapse and aggregation of long strands of glycine than the formation of H-bonds.
“Given that the backbone is a feature of all proteins, CO interactions may also play a role in proteins of nontrivial sequence where structure is eventually determined by interior packing and the stabilizing effects of H-bonds and CO-CO interactions,” he concluded.
The research was enabled by an allocation of compute time on Stampede through the Extreme Science and Engineering Discovery Environment (XSEDE) which is supported by the National Science Foundation.
Pettitt, a long-time champion of supercomputing, doesn’t only use TACC resources himself. He encourages other scholars, including his colleagues at the Sealy Center for Structural Biology and Molecular Biophysics, to use supercomputers as well.
“Advanced computing is important for data analysis and data refinement from experiments, X-ray and electron microscopy, and informatics,” he says. “All of these problems have big data processing issues that can be addressed using advanced computing.”
When it comes to uncovering the mysteries of biology on the tiniest scales, nothing quite beats a giant supercomputer.




Leave a Reply