Sign up for Scientific Inquirer’s Steady State Newsletter for the week’s top stories, exclusive interviews, and weekly giveaways. Plenty of value added but without the tax. http://bit.ly/2VEF06u
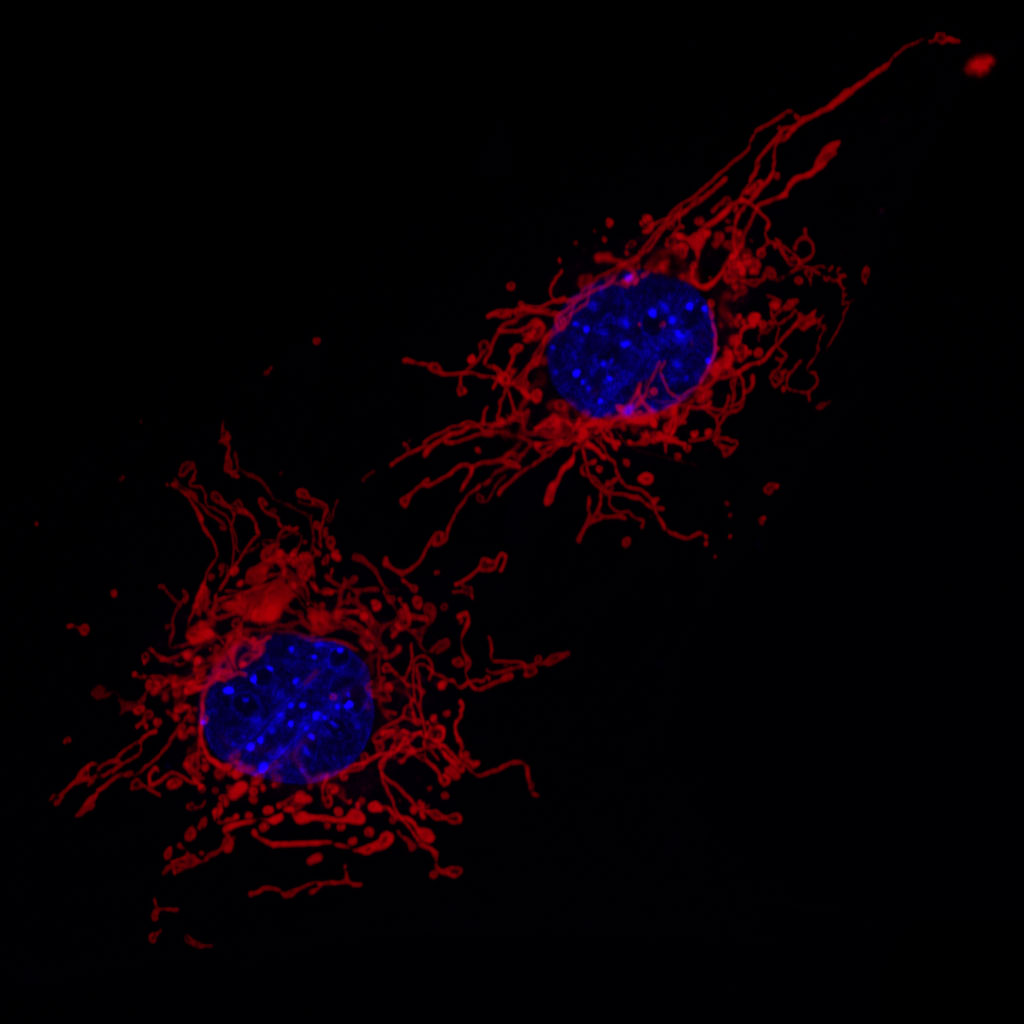
For the first time, using a mouse model of Alzheimer’s disease, scientists have documented a link between raised levels of calcium in mitochondria and neuronal death in the living brain. This relationship was previously documented in cell culture, but seeing this phenomenon in living mice makes it more likely that this occurs in people also and could point to a new target for Alzheimer’s disease.
“We were able to show mitochondrial calcium dysregulation in the neurons of living mice with Alzheimer’s-like symptoms using cutting edge live imaging techniques,” says the lead author of the paper, Maria Calvo-Rodriguez, PhD. The senior author is Brian J. Bacskai, PhD. They are both from the Department of Neurology at Massachusetts General Hospital.
Their collaborators included researchers from Harvard School of Public Health and the School of Medicine at Instituto de Investigacion Biomedica de Cadiz (INIBICA) in Spain. This study was recently published in Nature Communications.
One of the defining hallmarks of Alzheimer’s disease is the deposition of amyloid beta plaques and loss of neurons. The accumulation of amyloid beta has long been thought to be a trigger of the disease, but the exact means by which neurons die in Alzheimer’s remain a mystery, and the amyloid beta theory has become controversial because so many drug candidates targeting amyloid beta have failed in clinical trials.
One of the effects of amyloid beta plaques is that they cause high calcium ion (Ca2+) levels in the brain cells. There is also evidence that, at least in cell culture, exposure to amyloid beta can raise Ca2+ levels within mitochondria and lead to neuronal death.
Mitochondria influence Ca2+ signaling inside neurons through the “mitochondrial calcium uniporter” that takes up Ca2+ into mitochondria. The investigation of this mechanism in living mice has been hampered by the lack of technologies sensitive enough to directly assess Ca2+ levels in mitochondria in the living brain.
To explore the relationship between Ca2+, mitochondria and neuronal death, Calvo-Rodriguez and her colleagues combined multiphoton-microscopy with a ratiometric Ca2+ indicator targeted to mitochondria to asses Ca2+ levels. They applied these technologies to examine the neurons of a transgenic mouse model of Alzheimer’s disease that develops amyloid plaques similar to those from human patients.
Their studies demonstrate that increased mitochondrial Ca2+ levels are associated with plaque deposition and neuronal death in this model, indicating that abnormal Ca2+ levels in mitochondria could play a role in neuronal cell death in Alzheimer’s disease.
Additionally, they observed that when soluble Aβ is applied to the healthy mouse brain Ca2+ concentration in mitochondria increases. That process can be prevented by blockage of the mitochondrial calcium uniporter with a drug. Soluble amyloid beta is a type of amyloid beta similar to that in the human Alzheimer’s brain.
“High calcium levels in the mitochondria cause oxidative stress, and the death of neurons via apoptosis,” says Calvo-Rodriguez. “We propose that by blocking the neuronal mitochondrial calcium uniporter we can prevent cell death and impact disease progression.” Their work suggests targeting calcium entry to the mitochondria could be a promising new therapeutic approach in Alzheimer’s disease.
IMAGE SOURCE: Creative Commons
Words matter. Images matter. The Scientific Inquirer needs your support. Help us pay our contributors for their hard work. Visit our Patreon page and discover ways that you can make a difference. http://bit.ly/2jjiagi


Leave a Reply