
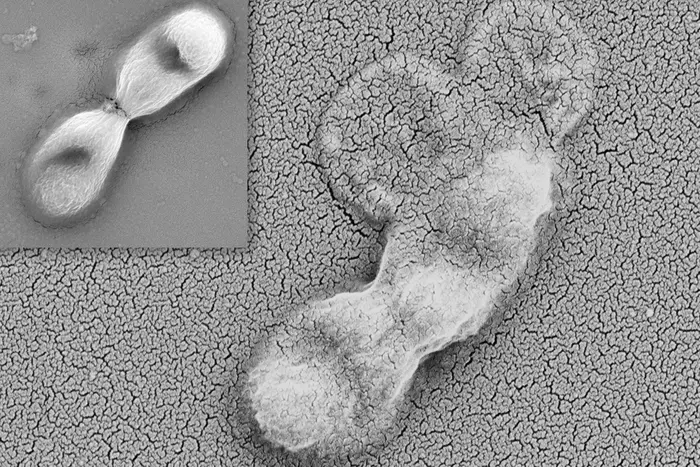
Like a spider trapping its prey, our immune system cells cooperate to capture and “eat” bacteria.
The newly identified antibacterial mechanism, reported Sept. 10 in Science Advances, could inspire novel strategies for combating Staphylococcus aureus (staph) and other extracellular bacterial pathogens.
It was known that neutrophils — first responder immune cells that migrate to sites of infection — can self-destruct and release their protein and DNA contents to generate neutrophil extracellular traps (NETs). Now, Vanderbilt researchers led by postdoctoral fellow Andrew Monteith, PhD, have discovered that NETs boost the bacterial killing power of another type of immune cell: macrophages.
“Neutrophils produce the spider webs that immobilize the bacteria, and macrophages are the spiders that engulf and kill the bacteria,” said Eric Skaar, PhD, MPH, Ernest W. Goodpasture Professor of Pathology, Microbiology and Immunology and director of the Vanderbilt Institute for Infection, Immunology and Inflammation.
Staph bacteria — particularly antibiotic-resistant forms — are a leading cause of hospital-acquired infections, infectious heart disease and pus-forming skin and soft tissue infections.
Neutrophils and macrophages are both phagocytic cells known for ingesting bacteria and producing antimicrobial peptides, reactive oxygen species and other enzymes to fight infection. NET generation (NETosis), thought to be a form of programmed cell death, is a more recently discovered neutrophil antibacterial strategy, Skaar said. The released neutrophil DNA creates a sticky trap that is also studded with antimicrobial peptides.
Monteith and colleagues used neutrophils that undergo increased NETosis in animal and in vitro model systems to study the biological function of NETs. They found that increased NETosis did not provide a killing advantage to neutrophils in isolation. But when macrophages were present, NET formation enhanced macrophage antibacterial activity by increasing phagocytosis — of staph bacteria stuck in the NETs along with neutrophil antimicrobial peptides.
“The macrophages end up with not only their own antibacterial arsenal, but also the neutrophils’ antibacterial arsenal, all in the same compartment killing the bacteria,” Skaar said.
Increased NETosis also boosted macrophage killing of other bacterial pathogens including Streptococcus pneumoniae and Pseudomonas aeruginosa. The findings suggest that neutrophil/NET-macrophage cooperation is a broadly used immune defense mechanism.
The researchers also showed that elimination of a staph nuclease enzyme that cuts DNA makes the bacteria even more sensitive to NET-macrophage killing.
“It seems as if extracellular pathogens like staph have evolved secreted nucleases so they can cut their way out of these NETs — chop off the spider web and escape,” Skaar said.
Blocking the nuclease would make the pathogens more susceptible to NET-mediated killing and may be a good antibacterial treatment strategy. This type of “anti-virulence” approach would allow phagocytic and other immune cells to do their jobs and kill the bacteria, Skaar said.
“Scientists are excited about the idea of anti-virulence strategies, because we know a lot about bacterial virulence mechanisms and can come up with creative ways to inhibit them,” he said. Current pharmaceutical efforts, however, focus on drugs that directly kill bacteria rather than blunting their virulence.
Monteith, Skaar and colleagues are continuing to explore questions of NETosis, including how and when neutrophils opt for this form of cell death. They are also interested in how individual differences in NETosis — perhaps because of genetic variation or disease states — affect infection. In people with certain autoimmune conditions, for example, reduced NETosis may increase susceptibility to staph infections.
Other authors of the Science Advances paper include Jeanette Miller, Noel Maxwell, and Walter Chazin, PhD. The research was supported by the National Institutes of Health (grants AI069233, AI073843, AI101171, HL144081) and the Edward P. Evans Foundation.


Leave a Reply